scMultiMap for integrative analysis with GWAS results
Chang Su
2025-06-12
GWAS.RmdThis vignette demonstrates how to apply scMultiMap to map associated peak-gene pairs and integrate the results with genome-wide association studies (GWAS) to elucidate the regulatory functions of GWAS variants in disease-associated cell types. As an example, we will reproduce the results from the scMultiMap manuscript, specifically those presented in the section ‘scMultiMap mapped GWAS variants of Alzheimer’s disease to target genes in microglia.’ At the end of this vignette, we will generate Figures 4d and 4e from the manuscript.
library(scMultiMap)
library(Signac)
library(Seurat)
library(ggplot2)
library(openxlsx)
library(GenomicRanges)Motivation
Over 75 genetic loci have been associated with the risk of developing
Alzheimer’s disease (AD) (Bellenguez, C.
et al.), yet their regulatory functions and relevant cellular
contexts remain unclear. Among brain cell types, microglia—the brain’s
innate immune cells—exhibit the highest enrichment of genetic
association with AD in their cis-regulatory elements (Nott, A. et
al., Gjoneska,
E. et al.). Therefore, this analysis focuses on microglia and uses
scMultiMap to identify candidate pairs of cis-regulatory
elements and their target genes. As two examples, we examine how
scMultiMap results can elucidate the regulatory function of
fine-mapped AD variants in microglia at two loci: PICALM and INPP5D.
Run scMultiMap
Note: this vignette use the same dataset as in vignette: scMultiMap for disease-control
studies. Please refer to it for details on pre-processing data and
running scMultiMap.
# set it to the directory where data are saved
data_dir <- '../../../data'
# load AD Multiome data
obj <- readRDS(sprintf('%s/processed_AD_DLPFC_15.rds', data_dir))
# subset to microglia
ct <- 'Microglia'
ct_obj <- subset(x = obj, subset = predicted.id == ct)In this analysis, we study the top 5000 highly expressed genes and top 50000 highly accessible peaks in microglia, and use a cis-region of width 1Mb to define candidate peak-gene pairs.
pairs_df <- get_top_peak_gene_pairs(ct_control,
gene_top=5000, peak_top=50000,
distance = 5e+5,
gene_assay = 'RNA', # name of the gene assay
peak_assay = 'peaks') # name of the peak assay
# In the paper, we used control microglia to define peak-gene pairs. (`ct_control`)
# One can also use all microglia.Here, we generated 135401 candidate peak-gene pairs based on the criterion above. The users can also customize the peak-gene pairs based on your analysis goal.
Next, we estimate peak-gene associations in microglia from the
control and the Alzheimer’s disease groups, respectively. Note that
there are multiple biological samples in this dataset, so
scMultiMap should be run with bsample to avoid
spurious associations due to heterogeneity across biological samples
(see Methods in scMultiMap’s manuscript
for more details).
# multiple biological samples
table(ct_obj$id)
#>
#> 1224 1230 1238 3329 3586 4305 4313 4443
#> 201 161 283 211 152 347 17 171
#> 4481 4482 4627 HCT17HEX HCTZZT NT1261 NT1271
#> 233 415 241 264 26 192 265
# control
control_res <- scMultiMap(ct_control,
pairs_df,
bsample = 'id',
gene_assay = 'RNA',
peak_assay = 'peaks')
#> Start step 1: IRLS
#> Start IRLS for RNA
#> Start IRLS for peaks
#> Start step 2: WLS
#> There are 4652 unique genes in the peak-gene pairs.
#> Warning in sqrt(deno): NaNs produced
#> scMultiMap elapsed time: 01:13 (mm:ss)
# disease
disease_res <- scMultiMap(ct_AD,
pairs_df,
bsample = 'id',
gene_assay = 'RNA',
peak_assay = 'peaks')
#> Start step 1: IRLS
#> Start IRLS for RNA
#> Start IRLS for peaks
#> Start step 2: WLS
#> There are 4652 unique genes in the peak-gene pairs.
#> scMultiMap elapsed time: 01:16 (mm:ss)What does “Warning in sqrt(deno): NaNs produced” mean?
This warning suggests that some peaks or genes have variance
estimated to be negative, causing numerical errors in calculating
correlations. This situation is typically rare and may happen when you
consider genes or peaks with extremely low abundance (e.g. top 5000
genes and top 50000 peaks here). The estimated correlations will be
internally set to 0 by scMultiMap.
Load results on fine-mapped AD GWAS variants
# AD GWAS variants fine-mapped and priortized based on eQTL & Hi-C
# "Seeding from PAINTOR or FGWAS 95% credible set variants (Sup- plementary Table 6b),
# we first included their linkage disequilibrium (LD) tags (defined as LD R2 > 0.8) in TOP-LD European (EUR)
# and then retained only those overlapping microglia ATAC–seq peaks that interact with promoters"
#
# Reference: https://www.nature.com/articles/s41588-023-01506-8
# Data source: supplementary table 6b
mic_fm_prior_variants_df <- read.xlsx(sprintf('%s/41588_2023_1506_MOESM3_ESM.xlsx', data_dir),
sheet = 'Supplementary Table 6c', startRow = 2)
mic_fm_prior_variants_df$chr <- paste0('chr', mic_fm_prior_variants_df$chr)
grange_mic_fm_prior <- GenomicRanges::makeGRangesFromDataFrame(df = mic_fm_prior_variants_df,
seqnames.field = 'chr',
start.field = 'pos.hg38',
end.field = 'pos.hg38')Visualize inferred peak-gene links and fine-mapped GWAS variants
PICALM locus (figure 4d)
# make grange objects that connect genes with peaks
gene_peak_link_gr_control <- make_gene_to_peak_link(control_res, 'PICALM', ct_control, 'peaks')
gene_peak_link_gr_AD <- make_gene_to_peak_link(disease_res, 'PICALM', ct_AD, 'peaks')
gene_peak_link_list <- list(AD = gene_peak_link_gr_AD, control = gene_peak_link_gr_control)
uniform_fs <- 20
# genomic region around PICALM locus
selected_range <- "chr11-85850000-86216290"
gene_plot <- AnnotationPlot(
object = ct_control,
region = selected_range
) + theme(text = element_text(size = uniform_fs))
link_plot_list <- list()
for(g in c('control','AD')){
Links(ct_obj) <- gene_peak_link_list[[g]][abs(gene_peak_link_list[[g]]$score) > 0.2]
link_plot_list[[g]] <- LinkPlot(
object = ct_obj,
region = selected_range
) + scale_color_gradient2(low = "blue",
midpoint = 0,
mid = "white",
high = "red",
space="Lab",
limits = c(-1,1)) +
labs(color = 'correlation', y = sprintf('%s', g)) +
theme(text = element_text(size = uniform_fs))
}
#> Scale for colour is already present.
#> Adding another scale for colour, which will replace the existing scale.
#> Scale for colour is already present.
#> Adding another scale for colour, which will replace the existing scale.
expr_plot <- ExpressionPlot(
object = ct_obj,
features = "PICALM",
assay = "SCT",
group.by='Status'
) +
theme(text = element_text(size = uniform_fs))
# to see it on the plot
end(grange_mic_fm_prior) <- end(grange_mic_fm_prior) + 1000
DefaultAssay(ct_obj) <- 'peaks'
snp_plot <- PeakPlot(
ct_obj,
selected_range,
assay = NULL,
peaks = grange_mic_fm_prior,
group.by = NULL,
color = "black",
sep = c("-", "-")
) + labs(y = 'SNPs') +
theme(text = element_text(size = uniform_fs))
g <- CombineTracks(plotlist = list(gene_plot, snp_plot, link_plot_list[[1]], link_plot_list[[2]]),
expression.plot = expr_plot,
heights = c(2,1,2,2) * 5/7,
widths = c(6.5, 1))
g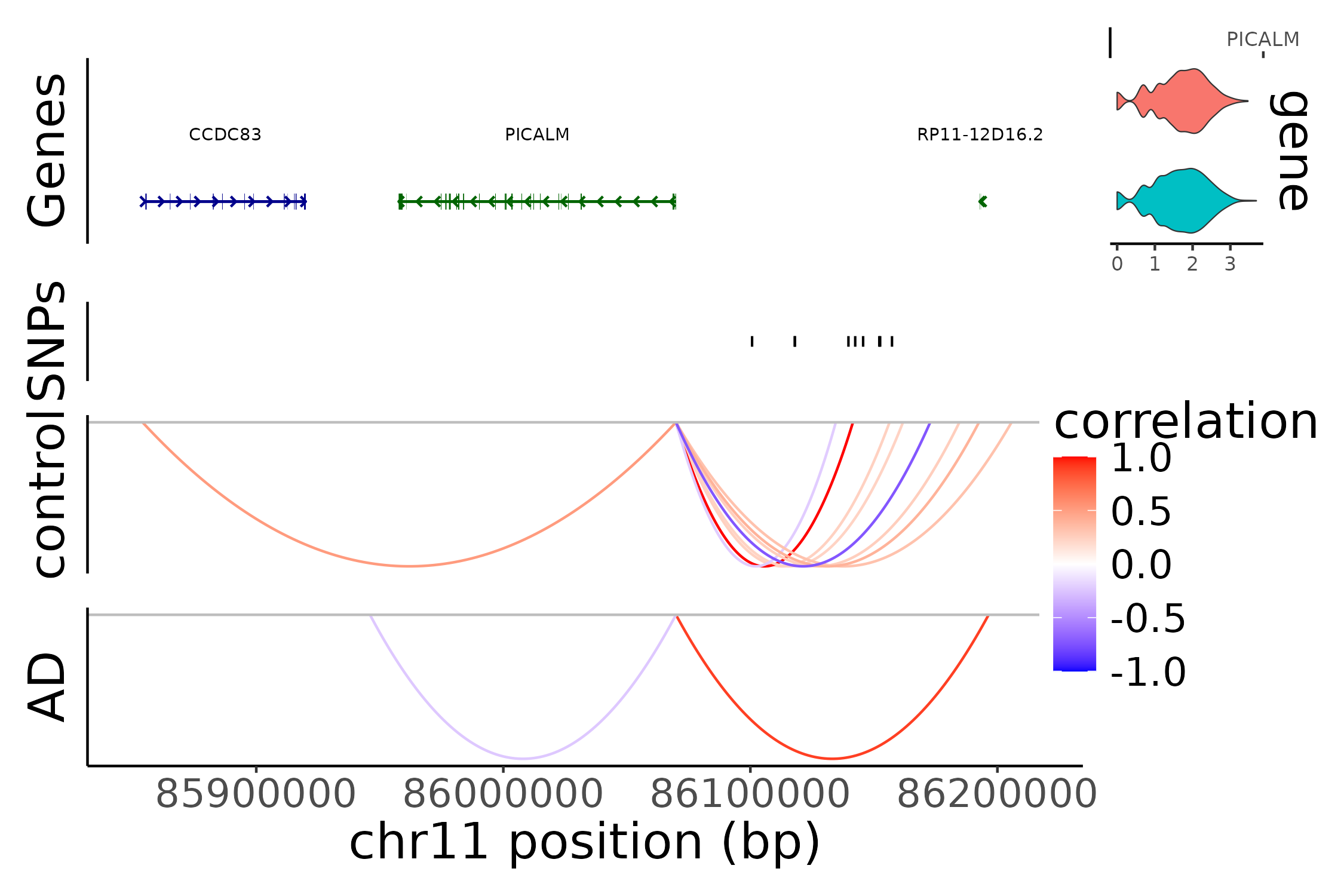
This reproduced Figure~4d in the scMultiMap’s manuscript.
INPP5D locus (figure 4e)
Repeat the analysis above for the INPP5D locus.
# make grange objects that connect genes with peaks
gene_peak_link_gr_control <- make_gene_to_peak_link(control_res, 'INPP5D', ct_control, 'peaks')
gene_peak_link_gr_AD <- make_gene_to_peak_link(disease_res, 'INPP5D', ct_AD, 'peaks')
gene_peak_link_list <- list(AD = gene_peak_link_gr_AD, control = gene_peak_link_gr_control)
uniform_fs <- 20
# genomic region around INPP5D locus
selected_range <- "chr2-232850000-233259353"
gene_plot <- AnnotationPlot(
object = ct_control,
region = selected_range
) + theme(text = element_text(size = uniform_fs))
link_plot_list <- list()
for(g in c('control','AD')){
Links(ct_obj) <- gene_peak_link_list[[g]][abs(gene_peak_link_list[[g]]$score) > 0.2]
link_plot_list[[g]] <- LinkPlot(
object = ct_obj,
region = selected_range
) + scale_color_gradient2(low = "blue",
midpoint = 0,
mid = "white",
high = "red",
space="Lab",
limits = c(-1,1)) +
labs(color = 'correlation', y = sprintf('%s', g)) +
theme(text = element_text(size = uniform_fs))
}
#> Scale for colour is already present.
#> Adding another scale for colour, which will replace the existing scale.
#> Scale for colour is already present.
#> Adding another scale for colour, which will replace the existing scale.
expr_plot <- ExpressionPlot(
object = ct_obj,
features = "INPP5D",
assay = "SCT",
group.by='Status'
) +
theme(text = element_text(size = uniform_fs))
# to see it on the plot
end(grange_mic_fm_prior) <- end(grange_mic_fm_prior) + 1000
DefaultAssay(ct_obj) <- 'peaks'
snp_plot <- PeakPlot(
ct_obj,
selected_range,
assay = NULL,
peaks = grange_mic_fm_prior,
group.by = NULL,
color = "black",
sep = c("-", "-")
) + labs(y = 'SNPs') +
theme(text = element_text(size = uniform_fs))
g <- CombineTracks(plotlist = list(gene_plot, snp_plot, link_plot_list[[1]], link_plot_list[[2]]),
expression.plot = expr_plot,
heights = c(2,1,2,2) * 5/7,
widths = c(6.5, 1))
g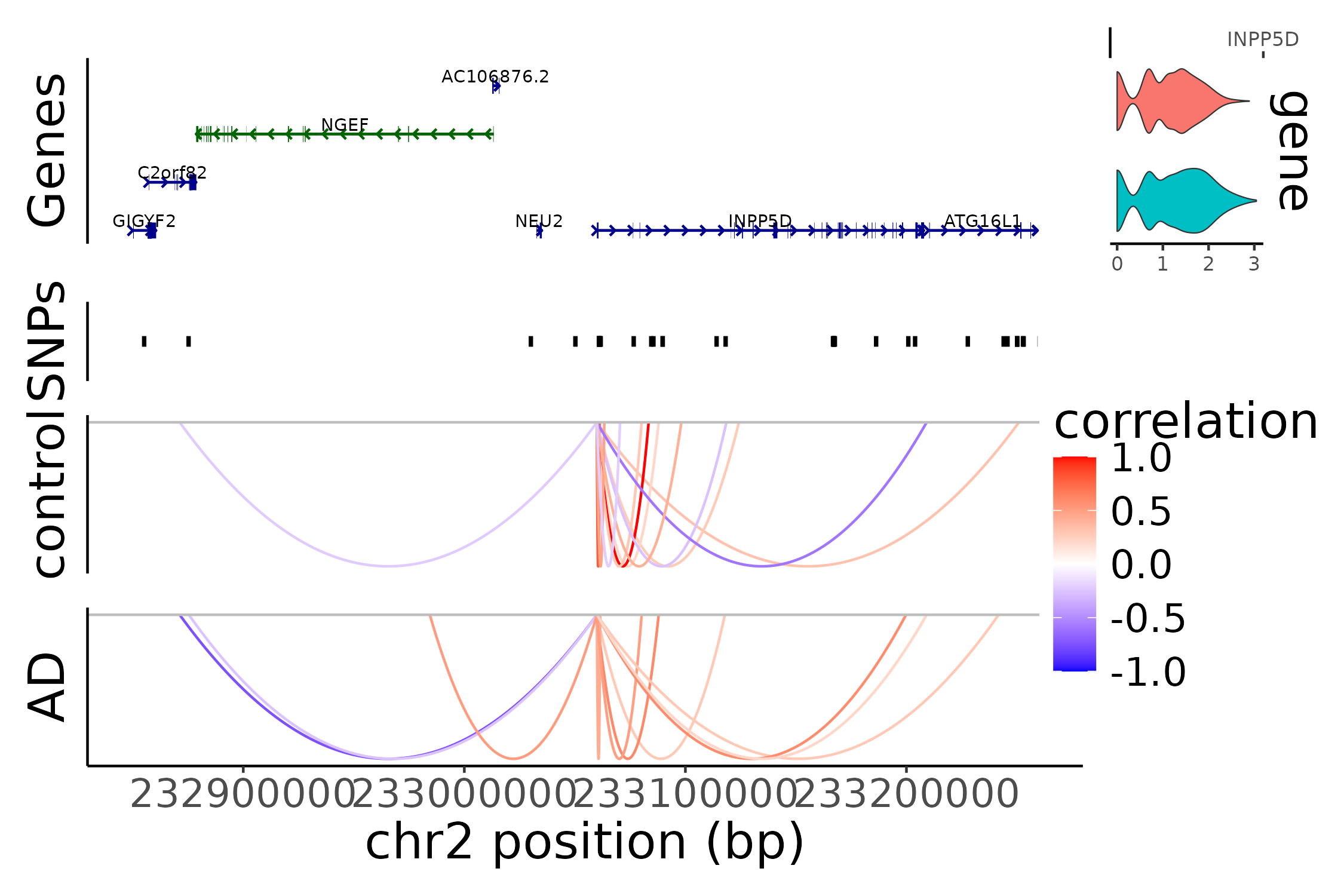
This reproduced Figure~4e in the scMultiMap’s manuscript.